Jennifer Gerton and Karen Miga had been longtime colleagues and shared an interest in hard-to-sequence regions of the genome for much of their careers.
“Karen wanted to pick a uniparental cell line with a stable genome for sequencing, to help complete the human genome, but she didn’t have access to her lab. She asked if we could evaluate some cell lines for her,” recalled Gerton, of how she initially became involved in the consortium.
Tamara Potapova, PhD, Leonardo Gomes de Lima, PhD, and Matt Borchers
Miga and colleagues initially sought a source of human genome
material that contained identical pairs of chromosomes from only one
parent, to avoid the added complication of assembling both a maternal
and paternal genome. This condition was satisfied by a human cell line
derived from a rare occurrence called a hydatidiform mole.
“The hydatidiform mole forms when something goes wrong during
conception,” explained Tamara Potapova, PhD, a research specialist II in
the Gerton Lab. “The egg’s (maternal) genome is lost, and the paternal
genome gets duplicated, resulting in a genome with mostly identical
pairs of chromosomes.”
Because hydatidiform mole tissue can easily become aneuploid (having
extra or missing chromosomes), which would pose a problem for
researchers having to contend with variable copies of certain regions,
the need to identify a stable cell line, with a normal number of
chromosomes, was urgent.
“Tamara’s expertise in imaging chromosomes has been a huge asset for
the project,” Gerton said. “She has a talent for culturing and
evaluating cell lines that are very difficult to work with.”
During the early stages of the project, and for the main Science paper, Potapova helped identify the CHM13 cell line as the most stable of the hydatidiform mole cell lines.
“At that point, Tamara became a cytogeneticist for the whole
project,” Gerton said. “When it comes to the ground truth of assembled
sequence, does it match what we see in the chromosomes by microscopy?
Tamara is the person we go to when we ask those questions. The beautiful
picture of spectral karyotyping, identifying all the chromosomes, on
the T2T website—that’s hers.”
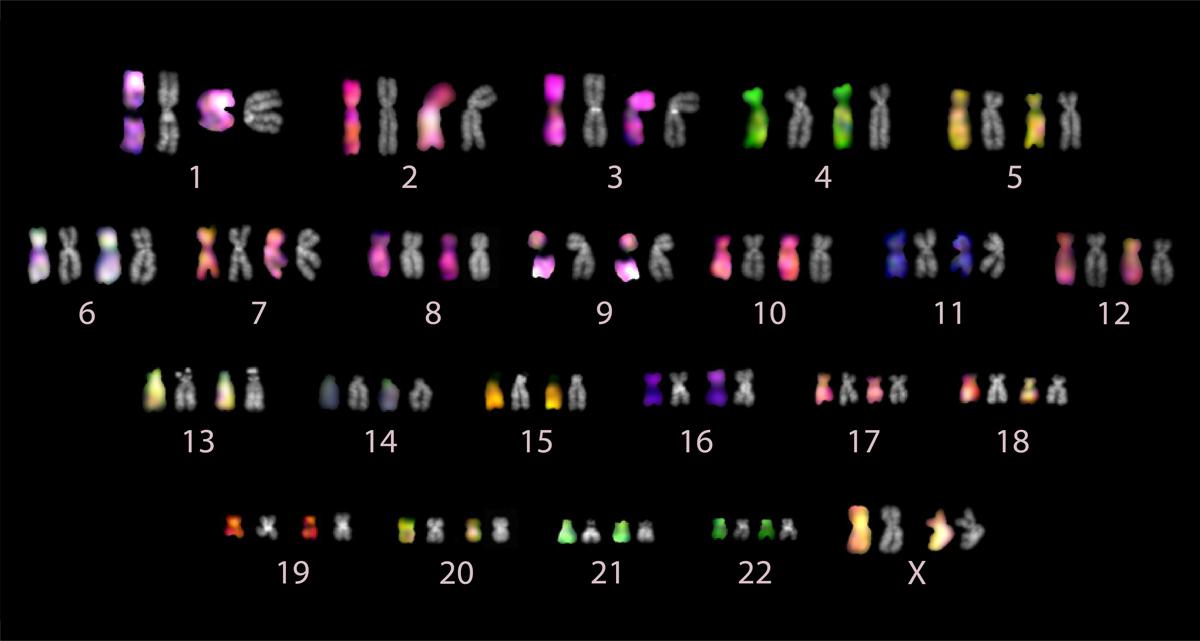
An ordered collection of the pairs of chromosomes analyzed in the T2T complete human genome sequencing project. Image courtesy of the Gerton Lab.
Potapova studies nucleolar organizing regions, which organize the
nucleolus, a specialized compartment within the cell’s nucleus. These
genetic regions consist primarily of ribosomal DNA, which has unique
behavior relative to most other coding regions of DNA.
Because ribosomal DNA genes are repetitive and are present in
multiple, nearly identical copies, assembling these regions of the human
genome was previously impossible. New sequencing technologies that
produced long and highly accurate stretches or "reads" of DNA made it
possible for Adam Phillippy’s group to find a path through this "dark
matter."
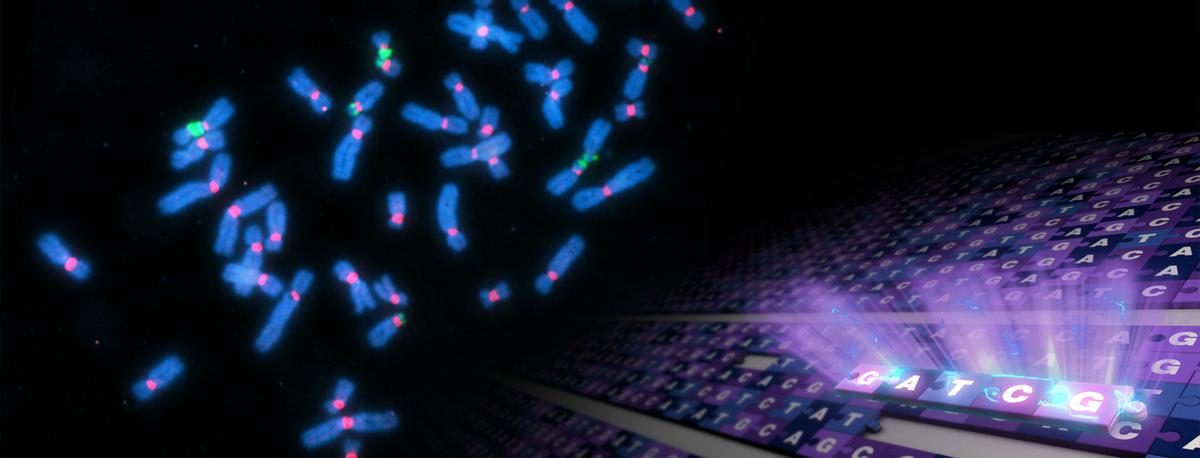
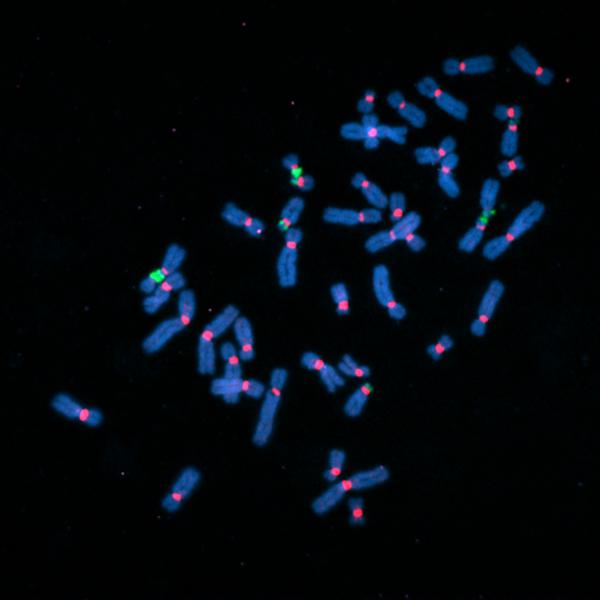
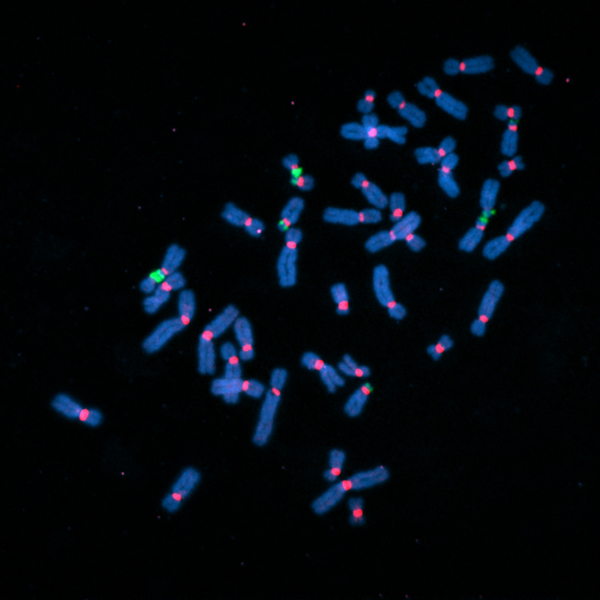
Fluorescently labeled human chromosomes (blue) showing attachment points called centromeres (red) and regions of ribosomal DNA (green). Many of the sequencing gaps were located in these regions. Image courtesy of the Gerton Lab.
“Adam’s group used this long-read sequencing method because a single
‘read’ could span multiple repeats and also the neighboring regions.
Untangling these reads so they could assign them to a particular
chromosome was a very complex process,” said Potapova.
“A brilliant computer scientist in my lab, Sergey Nurk, PhD,
developed new methods to squeeze every last bit of information out the
latest sequencing data,” said Phillippy. “With his tools, it was like
putting on a new pair of glasses. All of sudden we could see every
region of the genome with unprecedented clarity.”
“We wanted to know with high certainty how many ribosomal DNA repeats
each chromosome had in order to fill the gaps with the correct number
of copies. That question was very hard to approach even by modern
sequencing methods. But we could estimate the number of ribosomal DNA
repeats on each individual acrocentric chromosome by fluorescence
microscopy,” said Potapova.
For the paper, “we made chromosome preparations and marked the
ribosomal DNA with fluorescent labels. We knew the total copy number of
the repeats from sequencing and a special PCR technology called droplet
digital PCR, and we could measure the fluorescence intensity of all
ribosomal DNA locations in a chromosome preparation. From that, we could
calculate the fraction of the total fluorescent signal that was present
on every acrocentric chromosome and convert that to the number of
copies of the repeats.”
“Jay Unruh, PhD, who is director of scientific data at the Stowers
Institute, was very helpful with analyzing this imaging data, which is
not trivial,” said Potapova. “We benefited greatly from all the advice
and feedback that Jay and the Microscopy Center team contributed to our work on an ongoing basis.”
This unprecedented resolution enables scientists to ask new questions
about ribosomal DNA, and more generally, acrocentric chromosomes.
Scientists can ask how these chromosomal regions are inherited from
parent to child and how they organize chromosomes in three dimensions.
Because ribosomal DNA is crucial for cellular function, this information
opens new doors for understanding how cells develop into tissues, and
how health and disease depend on ribosomal DNA.