Newsletter


31 July 2026
Stowers News - July 2026
Read Article
News
Recent studies link severe craniofacial disorders to disruptions during development
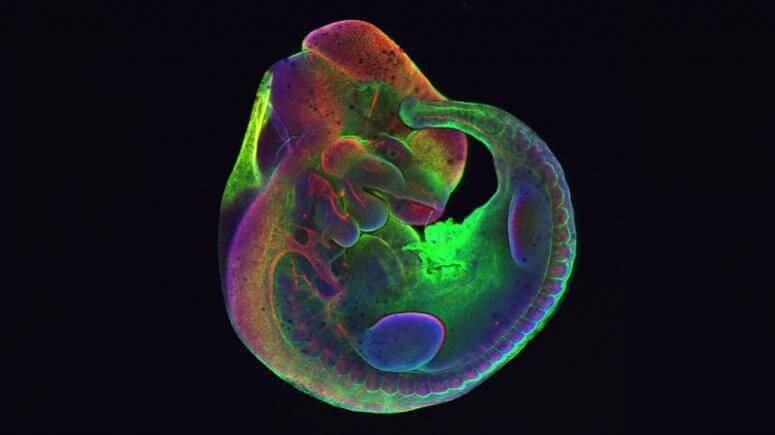

Fluorescent microscopy image of a mouse embryo. Red staining indicates peripheral neurons and nerves derived from neural crest cells.
By Rachel Scanza, PhD
If the eyes are the window to the soul, the face is what we see upon entering - a gateway to emotion, self-identity, how we are perceived in society, and how we communicate with others. During the last two and a half years, the pandemic required not only social distancing but also, quite literally, masking the part of our body fundamental to our social interactions. The impact was and continues to be for many, a sense of isolation. For people born with facial differences, that inability or difficulty in communicating emotion, can be a lifelong struggle.
The development of the head and face is a complex process that commences during the first month of pregnancy. Ribosomes, the biological machines that produce proteins required for every cellular process, are manufactured from ribosomal RNA (rRNA) and a plethora of proteins. Disruptions in transcription of rRNA—the process where integral stretches of DNA are copied—can result in a host of defects that not only impact appearance, for example in the form of cleft lip, but can also cause severe developmental and neurological disorders.
Understanding the mechanisms that govern normal and abnormal craniofacial development may lead to therapies that are preventative, or at the very least, diminish the severity of these pathologies.
New research from the Stowers Institute for Medical Research has studied several genetic factors involved in rRNA transcription and ribosome biogenesis—the construction of ribosomes from rRNA and proteins—and discovered how mutations in these genes preferentially affect cells and tissues that form head and facial structures. Recent Stowers Graduate Karla Falcon, PhD, and Postdoctoral Researchers Kristin Watt, PhD, and Soma Dash, PhD, in the lab of Stowers Investigator Paul Trainor, PhD, published a study in PNAS on July 26, 2022, that specifically examines subunits of RNA Polymerase I, the enzyme responsible for rRNA transcription, and an associated gene, and how mutations in these genes result in severe craniofacial disorders like Treacher-Collins Syndrome and Acrofacial Dysostosis-Cincinnati Type.
“Every cell needs ribosomes in order to make proteins. Deficiencies in ribosome quantity or quality compromise the ability of a cell to make proteins, and those cells then cannot function properly,” said Trainor.
“The surprising thing about ribosome pathologies is that despite the global requirement for ribosomes in all cells, development of the head and face is often particularly affected,” said Watt.
Neural crest cells, which are similar to stem cells, rapidly form around three to four weeks into human gestation. This cell population detaches from the primitive central nervous system, increases in number via cell division, and migrates into the developing head as well as many other parts of the body.
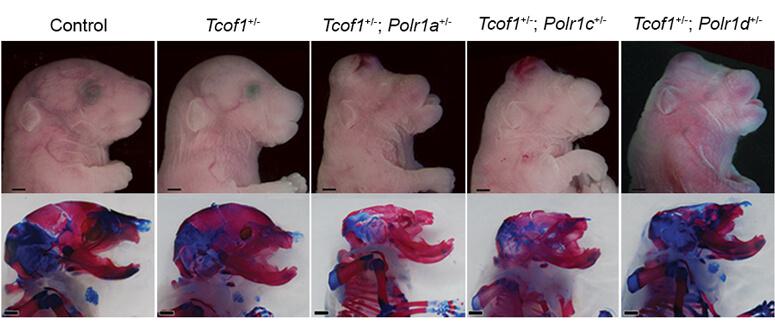

Mouse embryos at 18.5 days post-fertilization. Combinations of mutations in the RNA Polymerase I genes and associated gene factor Tcof1 increase the severity of craniofacial anomalies.
The detachment of neural crest cells occurs via a process called epithelial to mesenchymal transition—where static cells change their shape and acquire migratory properties—the subject of a recent review paper from the Trainor Lab. Once situated in the facial region, neural crest cells differentiate, or decide their cell fate, ultimately generating nearly all the of the craniofacial cartilage, bone and connective tissues.
The rapid proliferation and growth of neural crest cells in concert with epithelial to mesenchymal transition requires a disproportionate quantity of ribosomes and hence proteins compared to surrounding non-neural crest cells and tissues. One of the key steps of ribosome biogenesis—RNA Polymerase I-mediated rRNA transcription—is a rate-limiting step in the formation of ribosomes, and is highly active within neural crest cells.
“Large quantities of protein translation are needed for neural crest cell proliferation and survival,” said Dash.
By turning off genes responsible for rRNA transcription and ribosome formation in mice, the researchers uncovered a cellular imbalance between rRNA transcription and ribosomal proteins that greatly impacted neural crest cells.
This imbalance promotes neural crest cell death via increased concentrations of the tumor suppressor protein, p53, which causes the embryo to develop malformations of the head and face.
In normal neural crest cells, p53 levels are tightly controlled by the protein, Mdm2. However, when rRNA transcription is reduced, an imbalance arises where ribosomal proteins that normally form part of the ribosome, become free and instead bind to Mdm2 preventing its control of p53. This leads to increased levels of p53 that cause the newly formed neural crest cells to die.
“We have known for many years that problems in craniofacial development are often the result of neural crest cells dying before they can form the cartilage and bone structures of the head and face,” said Falcon.
“Our previous studies have shown that inhibiting p53 in Treacher-Collins Syndrome animal models can stop neural crest cells from dying and thus prevent craniofacial anomalies from occurring, but this made the animals much more susceptible to cancer,” said Watt.
“The significance of this study is that at the genetic, cellular, and biochemical level, we can now connect rRNA transcription and the ribosome biogenesis pathway in neural crest cells, to proper craniofacial development and to the pathogenesis of birth defects,” said Trainor. “We understand how each step in the pathway links to p53-dependent apoptosis.”
Coauthors include Ruonan Zhao, Daisuke Sakai PhD, Emma L. Moore, Sharien Fitriasari, Melissa Childers, Mihaela E. Sardia, Selene Swanson, Dai Tsuchiya, Jay Unruh, PhD, George Bugarinovic, Lin Li, Rita Shiang, PhD, Annita Achilleos, PhD, Jill Dixon, PhD, and Michael Dixon, PhD.
Studying multiple research organisms is key
The study of rare diseases is challenging but deserves no less attention than common diseases. However, a universal mechanism to explain rare birth defects simply doesn’t exist and developing preventative therapies therefore requires a thorough understanding of the cellular and genetic mechanisms responsible for each individual disorder, which is derived from our knowledge of normal craniofacial development in many different animal species.
For example, in another recent study published in Development on June 28, 2022, Dash in the Trainor Lab investigated the function of Nucleolin in neural crest cells and craniofacial development in zebrafish. Mutations in Nucleolin disrupt the differentiation of neural crest cells resulting in craniofacial anomalies resembling Treacher-Collins Syndrome. Dash discovered that Nucleolin, among its many functions, regulates signaling pathways that are important for neural crest cell differentiation into bone and cartilage.
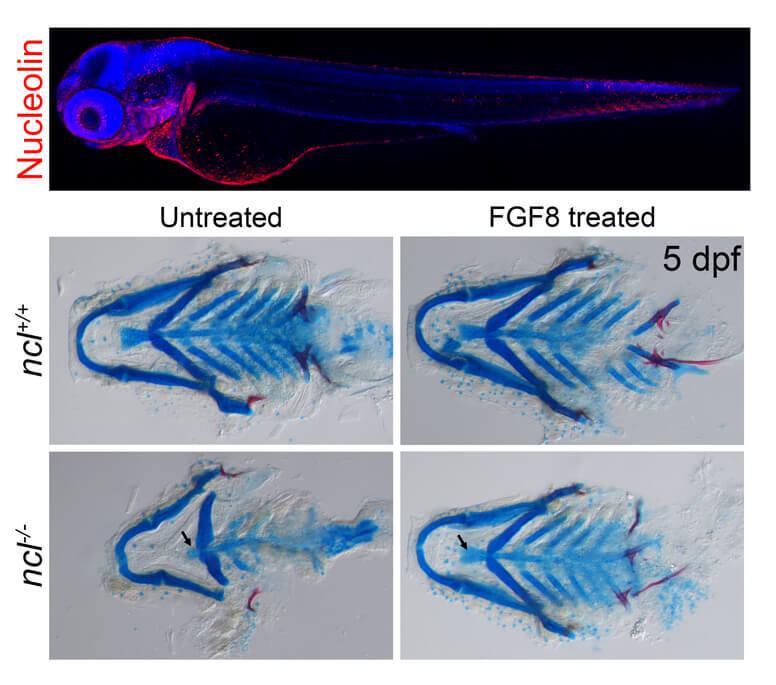

Zebrafish embryos have higher expression of Nucleolin (top, in red) in the craniofacial region at three days post-fertilization. Cranioskeletal rescue at five days post-fertilization with FGF8 protein treatment in nucleolin mutant embryos (bottom, right).
“By identifying the biochemical pathway that was involved, we were able to bypass the problem by providing the zebrafish embryos with the specific proteins they needed for proper differentiation,” said Dash.
Using research organisms such as mice and zebrafish to understand the basis for birth defect disorders can lead to discoveries on how to prevent them. “If it’s successful in animal models, it may give people hope that something can be done in the future,” said Trainor.
This work was funded by the Kirschstein-NRSA F31 predoctoral fellowship (awards DE027860, DE023017) from the National Institute for Dental and Craniofacial Research of the National Institutes of Health, K99 Pathway to independence awards (DE030971 and DE030972), the American Association for Anatomy Post-Doctoral Fellowship, and from institution support from the Stowers Institute for Medical Research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Press Release


30 July 2026
Charles McAnany, Ph.D., uses machine learning to help decode how DNA controls gene activity and reveal hidden patterns in the genome. He will now enhance AI accessibility and implementation across the Institute’s 24 research programs.
Read Article
News
30 July 2026
Promoted to Investigator, Randal Halfmann, Ph.D., has built a research program around the molecular events that help cells adapt, defend themselves, and sometimes begin a path toward disease — and there are plenty of questions he still wants to ask.
Read Article


