News


03 August 2026
Meet the Summer Scholar Uncovering How Bacteria Communicate
Jimuel Clarence Malimban is gaining hands-on research experience using genetic tools to better understand bacterial behavior
Read Article
News
New study shows how we can better learn our genome’s hidden grammar, potentially paving the way for personalized medicine.


This image, selected for the cover of Cell Genomics, draws parallels between the ancient Oracle of Delphi and modern neural networks trained to predict genomics data from DNA sequence. Just like the Oracle’s prophecies required the Pythia’s special insights, today's sequence models require specialized tools and knowledge of transcription to uncover the molecular mechanisms underlying the learned sequence rules. The embryo on the right represents the model training, while on the left, it is the Pythia’s subject of inquiry. The new study interprets sequence models to decipher how transcription factors downstream of Hippo signaling bind DNA and shape gene expression in trophoblast cells from early mouse embryos.
Cells communicate with each other with a relatively limited repertoire of signals, yet the responses those signals elicit are vast. This is because the “language” encoded in our genome’s DNA is complicated and difficult to decipher. New research from the Stowers Institute for Medical Research reveals that a cell’s response to a signal is encoded in the DNA sequence and that the underlying grammar can be deciphered with artificial intelligence (AI). These tools learn the sequence rules for how genes are regulated and have the potential to revolutionize personalized medicine.
Led by Khyati Dalal, Ph.D., a former predoctoral researcher in the lab of Stower Investigator Julia Zeitlinger, Ph.D., the team sought to understand a simple yet surprisingly complex question—how do embryonic cells know how to respond to developmental cues? These “cues” called signaling pathways are rapid-response signals that activate regulatory proteins known as transcription factors and interact with cell-specific proteins to drive cell specialization. The new study, published in Cell Genomics on April 1, 2025, indicates that a specific cell type’s response to developmental signals is hardcoded within its DNA.
At the simplest level, DNA abides by two “codes”—the genetic code that instructs how proteins are made from genes and the regulatory code that determines when and in which cells this occurs. While all cells have the same DNA sequence, they have different transcription factors, and this means that different parts of the regulatory code are accessed and read by the cell.
“The regulatory code is why cells respond differently to signals,” said Zeitlinger. “Signaling pathways work together with the transcription factors of the regulatory code, and it is the combination that specifies which genes are turned on that give a cell a specific function.”
How machines can read flexible rules to find variability in surprising places
Hippo signaling, a key pathway controlling cell division during early development, derives its name from a very large mammal (the hippopotamus) to ensure organs do not grow to gargantuan sizes. But growth is not the only way by which cells respond to Hippo signaling. When a fertilized mouse egg starts to develop, the Hippo signaling pathway determines whether a cell becomes part of the trophoblast—the cells surrounding the embryo—or remains inside the fertilized egg to form the embryo. This makes it an ideal system to investigate how signaling pathways mediate cell type-specific responses.

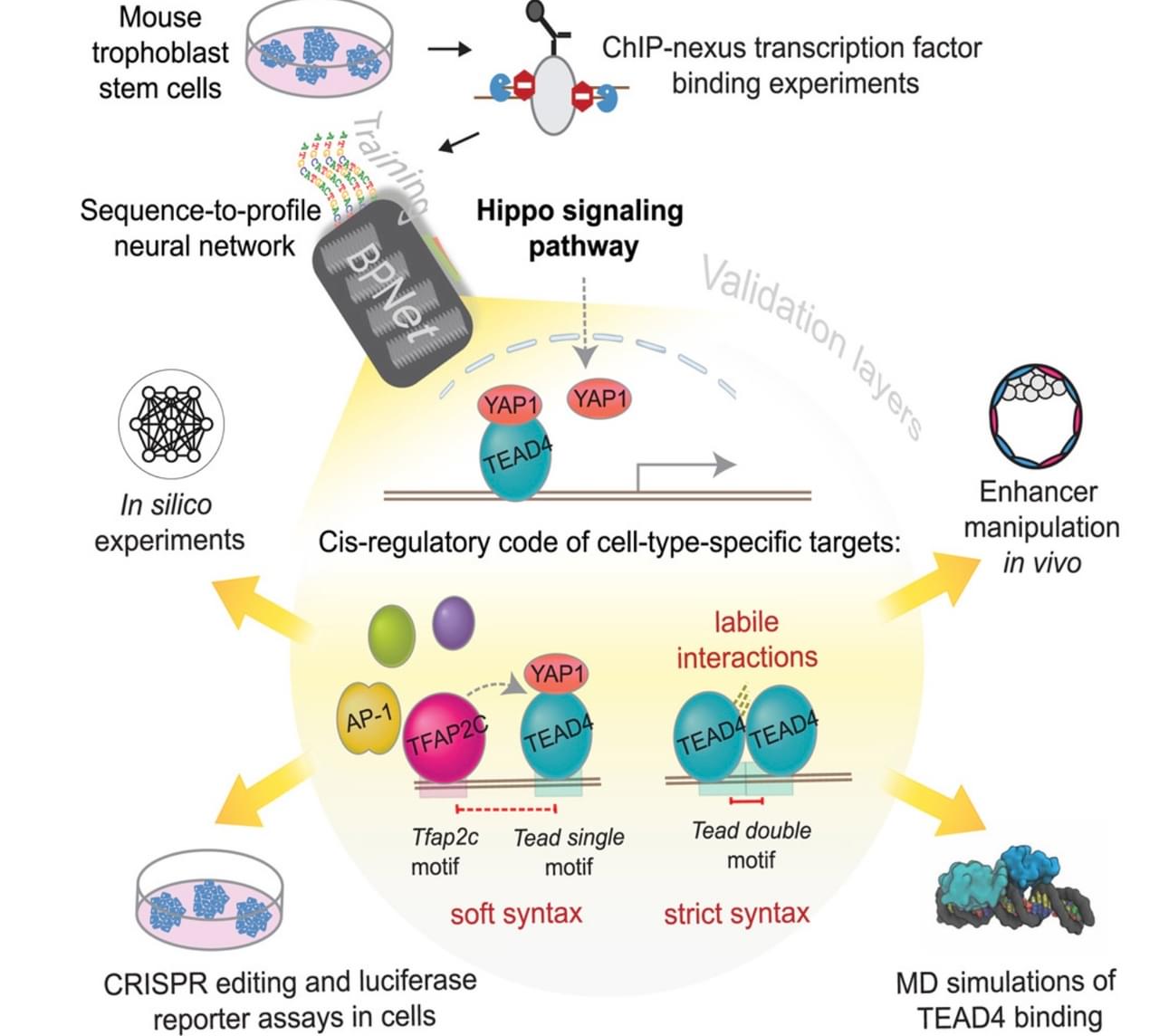
Graphical abstract depicting the methodology of deciphering how mouse trophoblast stem cells and the Hippo signaling pathway coordinate.
The team investigated how two transcription factors that belong to the Hippo signaling pathway, YAP1 and TEAD4, bind DNA together with transcription factors unique to mouse trophoblast stem cells to control which genes are turned on. The researchers utilized the AI tool BPNet, an interpretable deep learning framework that can both make predictions and can also explain how those predictions were made, to learn genome-wide relationships between DNA sequence patterns and transcription factor binding profiles.
“We reasoned that if the binding of YAP1 and TEAD4 is driven by the regulatory code, then the model should be able to learn their binding profiles from DNA sequence alone,” said Zeitlinger. “Moreover, analyzing the rules that the model learned should provide insights into exactly how the regulatory code is read.”
First, the researchers fed BPNet with real data showing where TEAD4, YAP1, and several other transcription factors are found in mouse trophoblast cells. After the model showed it could make accurate predictions from DNA sequence alone, they tested whether it could recognize patterns in new DNA sequences it hadn’t seen before—and it did.
“BPNet learned genome-wide rules that are predictive, showing that the information is encoded in the DNA sequence,” said Zeitlinger. “This is a step toward applying this approach more broadly to learn the regulatory code in the human genome by which signaling pathways instruct cells.”
“How exactly cells respond to signaling pathways has puzzled me since my Ph.D.,” said Zeitlinger. “These pathways are often the targets of therapeutic drugs, yet how different cell types respond is still poorly understood—finding a solution to this problem was very gratifying. Even more exciting was that we could extract the learned rules from the model, and these rules made sense and taught us something new about how transcription factors function.”
The team focused on two discoveries that emerged from the model and validated these findings in the lab. First, they characterized how YAP1 and TEAD4 work together with the trophoblast-specific transcription factor, TFAP2C. They found that when TFAP2C binds to DNA, this makes it easier for TEAD4 to bind to nearby regions, which in turn helps YAP1 connect to TEAD4 and activate genes.
“What was surprising was that this boost by TFAP2C is higher the closer it occurs to TEAD4,” said Zeitlinger. “Nevertheless, it is a flexible mechanism, which may explain how signaling pathways can receive regulatory input from a variety of transcription factors in different cell types.”
Second, the researchers uncovered that pairs of TEAD4 binding sequences—previously thought to be rare—have very strong effects and are much more common in the genome than expected. Although scientists had seen these double TEAD4 sites before, they hadn’t realized how important they are to the Hippo signaling pathway.
“The widespread double TEAD4 patterns had remained hidden because they don’t look very similar,” said Zeitlinger. “However, AI was able to recognize them because they have strong effects on binding.”
The rules for how DNA-protein interactions drive gene activation and cell fate specialization are highly complex. However, AI is proving to be an extraordinarily powerful tool that is transforming how biologists study gene regulation.
“Our work strongly supports the notion that the code of gene regulation can be deciphered,” said Zeitlinger. “If so, this would have huge implications for human health, not just for predicting drug targets, but also for helping identify disease susceptibilities and enabling personalized medicine.”
Additional authors include Charles McAnany, Ph.D., Melanie Weilert, Mary Cathleen McKinney, Ph.D., and Sabrina Krueger, Ph.D.
This work was funded by the National Human Genome Research Institute of the National Institutes of Health (NIH) (award: R01HG010211) and with institutional support from the Stowers Institute for Medical Research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
News
03 August 2026
Jimuel Clarence Malimban is gaining hands-on research experience using genetic tools to better understand bacterial behavior
Read Article
Press Release


30 July 2026
Charles McAnany, Ph.D., uses machine learning to help decode how DNA controls gene activity and reveal hidden patterns in the genome. He will now enhance AI accessibility and implementation across the Institute’s 24 research programs.
Read Article
News
30 July 2026
Promoted to Investigator, Randal Halfmann, Ph.D., has built a research program around the molecular events that help cells adapt, defend themselves, and sometimes begin a path toward disease — and there are plenty of questions he still wants to ask.
Read Article


