< All Technology Centers
Systems Mass Spectrometry
The Systems Mass Spectrometry team is dedicated to adopting, adapting, and developing leading-edge mass spectrometry-based multi-omics approaches to support a variety of research projects at the Institute.


The Systems Mass Spectrometry Center is hiring!
Overview of Services
Research Services
The Systems Mass Spectrometry team is a highly trained group of technical experts committed to collaborative research efforts that investigate large-scale sets of proteins or small molecules, such as metabolites and lipids, extracted from cells and/or spatially-resolved within tissues.
Current mass spectrometry analysis workflows fall into two main categories depending on the analytes to be characterized.
Protein-centric analyses include:
- Discovery/Shotgun Proteomics | To detect, identify, and quantify, in a systematic, large-scale, and high-throughput manner, the proteins in a sample, as well as their post-translational modifications and interaction networks. Discovery proteomics approaches currently in development are centered around single cell or low input sample size, such as laser captured microdissections or purified exosomes.
- Structural Proteomics | To characterize different structural features of proteins within purified complexes or on a proteome scale.
- Targeted MS Assays | To specifically detect and quantify representative peptides from preselected proteins of interest and/or their post-translational modifications.
Small molecule analyses include:
- Metabolomics | To detect, identify, and quantify the classes of metabolites that are amenable to be characterized by mass spectrometry.
- Lipidomics | To study cellular lipids on a large scale.
- Epitranscriptomics | To quantify global levels of RNA modifications through the targeted detection of modified ribonucleosides and their isomers..
Analytes of interest may be introduced in mass spectrometers:
- From solutions | by coupling liquid chromatography (LC) separation with electrospray ionization.
- From tissue sections or plated cells | via Matrix-Assisted Laser Desorption/Ionization, which allows to not only identify and quantify analytes but also to resolve them in space, supporting the Spatial Omics analysis pipeline.


Technology
Tandem mass spectrometers (MS) are the main instruments supporting analyte characterization workflows. They are either coupled to nanoflow microcapillary liquid chromatography (LC) and/or integrated with MALDI (Matrix-Assisted Laser Desorption/Ionization) target plates as ion sources.
- 1x | Thermo Scientific LTQ Linear Ion Trap · custom-made nano-electrospray ionization source (nano-ESI) · Agilent 1100 quaternary HPLC
- 2x | Thermo Scientific Velos-Orbitrap · custom nano-ESI · Agilent 1260 quaternary HPLC
- 1x | Thermo Scientific Q-Exactive+ · Nanospray Flex Ion Source · Dionex UltiMate 3000 RSCLnano System · Variable Wavelength Detector (VWD)
- 1x | Thermo Scientific Orbitrap Fusion Lumos · FAIMS (high-Field Asymmetric waveform Ion Mobility Spectrometry) Pro interface · Nanospray Flex Ion Source ·Dionex UltiMate 3000 RSCLnano System · VWD
- 1x | Thermo Scientific Orbitrap Eclipse · FAIMS Pro Interface · Nanospray Flex Ion Source · Dionex UltiMate 3000 RSCLnano
- 1x | Bruker timsTOF FleX MALDI2 · standard electrospray ionization (ESI) interface ·CaptiveSpray source · Vacuum Insulated Probe Heated Electrospray Ion Source (VIP HESI) · nanoElute UHPLC




Software & Computing
Complex mass spectrometry data is processed and analyzed using a variety of software that are run on Windows workstations, GPU appliances, or Linux computational servers and cluster. In-depth data analysis is performed by core scientists and/or by data analysts embedded in the group.
Software
- For peptide identification from tandem mass spectrometry datasets using protein sequence databases and/or spectral libraries:
- ProLuCID v.1.3.3 | freeware from the Yates lab at the Scripps Research Institute
- Proteome Discoverer 2.5 with the SEQUEST-HT search engine and XlinkX 2.5 nodes | commercial (Thermo Scientific)
- PaSER (Parallel Database Search Engine in Real-time) 2022 with TIMS DIA-NN | commercial (Bruker)
For protein inference and/or quantitation:
- DTASelect/Contrast v. 1.9 | freeware from the Yates lab at the Scripps Research Institute
- Trans Proteomic Pipeline | freeware
- Percolator | freeware
- kite/NSAF7 v1.0.0 | in-house developed software suite
For small molecules identification and quantitation from compound libraries:
- Compound Discoverer 3.1 (Thermo Scientific)
- MetaboScape 3.0 (Bruker)
- Skyline
For analyzing mass spectrometry imaging data:
- SCiLS Lab (Bruker)
Computing
- 3x | High performance Windows workstations dedicated to Proteome Discoverer, Compound Discoverer, or ScILS Lab/Metaboscape
- 1x | GPU appliance connected to timsTOF FleX MALDI2 and running PaSER for real time search
- 1x | Linux computational server with 48 cores and 1.5TB memory exclusively dedicated to DTASelect/Contrast post-search data processing
- 1x | Linux computational cluster with 160 high-CPU nodes dedicated to ProLuCID searches




Team Contact
Laurence Florens
Director, Systems Mass Spectrometry
Stowers Institute for Medical Research
Laurence Florens established the Systems Mass Spectrometry Center, formerly known as Proteomics, at the Stowers Institute in July 2003 with Mike Washburn, and was named Director in 2022. Florens has developed, adopted, and applied advanced mass spectrometry-based approaches to drive biological discovery in a wide array of collaborative projects, leading to over 240 peer-reviewed publications with 46 different labs.


















Multi-Omics Technologies
The Proteomics Systems Mass Spectrometry team applies various liquid chromatography mass spectrometry (LC-MS) analysis pipelines to a wide variety of samples, from cells, tissues, whole animals, biofluids, to purified organelles and cellular compartments. Analytes not only include peptides and proteins, but also metabolites, lipids, glycans, and hydrolyzed RNAs. The recent deployment of mass spectrometry imaging adds a spatial dimension to these omics studies by mapping the distribution of these analytes within tissue sections.
Combining such complementary and large-scale data acquisition modes to characterize an array of analytes from the same samples, followed by machine learning analyses of the generated datasets, should enable drawing systems-wide pictures of complex biology.


Discovery Proteomics
Used to detect and quantify large numbers of proteins from a wide variety of biological samples. More in-depth analyses include detecting and quantifying post-translational modifications and/or deriving small-scale protein interaction networks. Discovery proteomics approaches currently in development are centered around single cell or low input sample size, such as laser captured microdissections or purified exosomes.
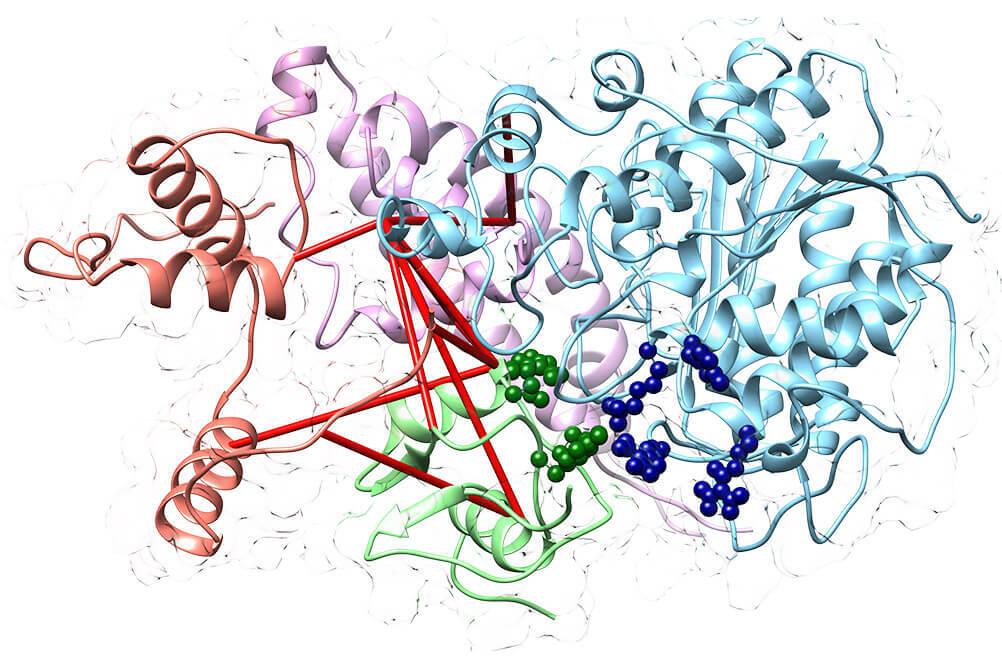



Structural Proteomics
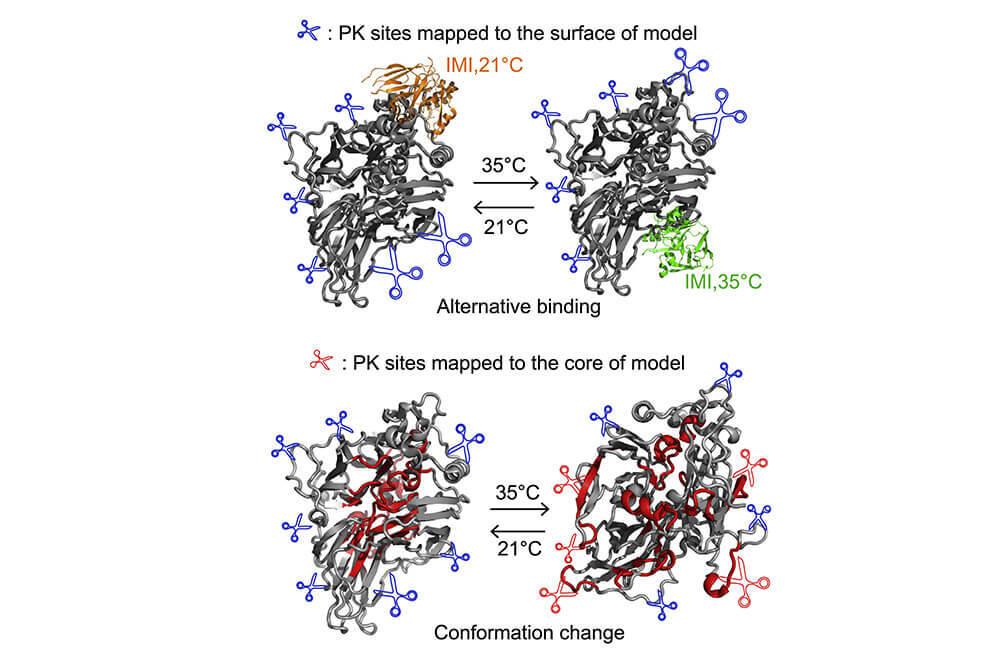
Studies different structural features of proteins within purified complexes, or on a proteome scale. Applications include investigating modularity within protein complexes, chemically crosslinking proteins within complexes to define interaction interfaces, assessing conformational changes with limited proteolysis or thermal proteome profiling, and characterizing intact proteins with top-down mass spectrometry.


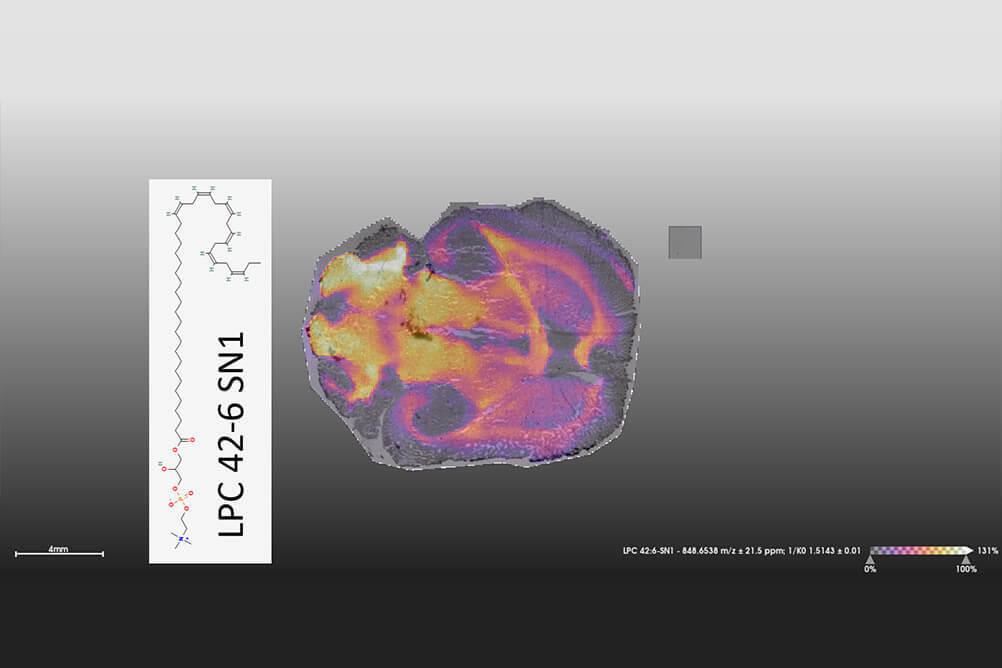



Spatial Omics
A new technology consisting of a timsTOF FleX MALDI2, which is a Dual MALDI Imaging and LC-MS platform. This system supports a "Spatial Omics" analysis pipeline from tissue sections and not only enables spatial resolution for proteins and metabolites, but also for lipids and glycans.
Multi-Omics Capabilities
Mass spectrometers are very versatile analytical tools that can be used to detect, identify, and quantify any type of analytes, as long as they can be ionized, and without the need for specific reagents such as antibodies. Specifically, mass spectrometry may be used to characterize key compound classes such as metabolites and lipids or to detect the occurrence of post-translational modifications like phosphorylation or glycation. Such biomolecules and post-translational events are active and dynamic participants in cell signaling and metabolism. However, conventional methods such as immunohistochemistry, fluorescence microscopy, or RNASeq are blind to them. Mass spectrometry-based analytical workflows may be implemented to study most molecular components of a sample, hence allowing for a systems-wide view of its biological context.
Targeted Proteomics
While Discovery Proteomics main goal is to detect and quantify as many proteins as possible from a sample, Targeted Proteomics is hypothesis driven. The mass spectrometry analysis of a preselected group of proteins allows more precise, quantitative, and sensitive data to be generated. Because of the limited number of ion species being targeted, these assays are more reproducible and quantitative and can dig deeper into a proteome dynamic range without enrichment.
Such targeted MS assays are complementary to discovery proteomics and usually implemented as an orthogonal method to more accurately validate qualitative observations or quantitative trends made in the shotgun phase of a project.
Metabolomics
We have deployed metabolomics analysis pipelines for the detection, identification, and quantitation of the classes of metabolites that are amenable to be characterized by liquid chromatography-based separations coupled to mass spectrometry.
Untargeted/Global LC-MS approaches are unbiased and comprehensive and help generate hypotheses. The data generated is mostly qualitative, at best semi-quantitative, and complex.
Targeted LC-MS approaches focus on a specific set of metabolites to test a hypothesis. The data generated is mostly quantitative if standard molecules are available to generate calibration curves.
Epitranscriptomics
Epitranscriptomics, also called “RNA epigenetics,” is a branch of epigenetics that refers to RNA editing and noncoding RNA regulations. Epitranscriptomics plays essential roles in alternative splicing, nuclear export, transcript stability, and translation of RNAs. RNA modifications are changes to the chemical composition of ribonucleic acid (RNA) molecules post-synthesis.
Many of the more than 170 modifications present in RNA have been known for decades, but only in the past several years have sufficiently sensitive tools been developed to identify and quantify these modifications in low-abundance RNA species such as mRNA.
One such sensitive and simple workflow we have adapted uses HPLC fractionation, a multistage higher-energy collisional dissociation fragmentation strategy, and Skyline to study the global differential expression level of modified ribonucleosides in various cellular states.
Published Source Code
The entire in-house software suite (kite) used for the processing and analysis of liquid chromatography tandem mass spectrometry data is available in Zenodo and at github. The suite includes scripts running in Windows, others in Linux.

RawDistiller
Extracts MS2 peak list into different file formats required by different search engines. Read Abstract on ACE Publications.
Kite
Job scheduler for MS2 data searching on Linux cluster that may be paused, stopped, and restarted at any time (i.e., impervious to hardware malfunction). Wraps jobs into different packages per the chosen search engine.
Swallow
Works with DTASelect v 1.9 to select PSMs at given specific spectra, peptides, and proteins FDRs.
Sandmartin
Works with Contrast to generate comparisons between multiple search results and report merge protein lists at given FDRs.
NSAF7
Generates quantitative contrast reports (calculating label-free dNXAF values for all detected proteins and PTMs) and calculates final FDRs. Read Abstract on ACS Publications.
Goose
Works with dtaselect2mzid and TPP to save the selected PSMs into a mzid file for complete submission to MassIVE.
News from the lab
News
06 February 2026
Stowers scientists discover a delivery system that helps fuel regeneration in planarians, solving a 25-year mystery
The new study reveals miniature “packages” help planarian cells share gene-silencing instructions across the body.
Read Article
News

24 September 2024
Stowers scientists look to zebrafish for potential insight into human fertility and development
Newly identified parental proteins present at fertilization reveal surprising levels of vertebrate developmental support
Read Article
Press Release
24 January 2023
Stowers scientists use cavefish to learn about metabolism and the evolutionary basis of being a couch potato
Researchers have discovered what prolonged physical inactivity may mean for humans many thousands of years down the road by studying cavefish.
Read Article
News

17 November 2022
#TechTalk: New Technology at Stowers helps identify biomolecules
Charles Banks is a scientist in the Stowers Institute’s Systems Mass Spectrometry Technology Center. We sat down with him to learn more about a new instrument Stowers members have access to—the timsTOF flex MALDI-2.
Read Article
Featured Publications
Dynamic regulation and requirement for ribosomal RNA transcription during mammalian development
Falcon KT, Watt KEN, Dash S, Zhao R, Sakai D, Moore EL, Fitriasari S, Childers M, Sardiu ME, Swanson S, Tsuchiya D, Unruh J, Bugarinovic G, Li L, Shiang R, Achilleos A, Dixon J, Dixon MJ, Trainor PA. Proc Natl Acad Sci U S A. 2022;119:e2116974119 doi: 10.1073/pnas.2116974119.
Mattingly M, Seidel C, Munoz S, Hao Y, Zhang Y, Wen Z, Florens L, Uhlmann F, Gerton JL. [published ahead of print June 3 2022]. Curr Biol. 2022;32.
Bhattacharya S, Wang S, Reddy D, Shen S, Zhang Y, Zhang N, Li H, Washburn MP, Florens L, Shi Y, Workman JL, Li F. Nat Commun. 2021;12:6452. doi: 6410.1038/s41467-41021-26799-41463.
Multiple roles for PARP1 in ALC1-dependent nucleosome remodeling
Ooi SK, Sato S, Tomomori-Sato C, Zhang Y, Wen Z, Banks CAS, Washburn MP, Unruh JR, Florens L, Conaway RC, Conaway JW. Proc Natl Acad Sci U S A. 2021;118:e2107277118. doi: 2107277110.2107271073/pnas.2107277118.
Proteome plasticity in response to persistent environmental change
Domnauer M, Zheng F, Li L, Zhang Y, Chang CE, Unruh JR, Conkright-Fincham J, McCroskey S, Florens L, Zhang Y, Seidel C, Fong B, Schilling B, Sharma R, Ramanathan A, Si K, Zhou C. Mol Cell. 2021;81:3294-3309 e3212.
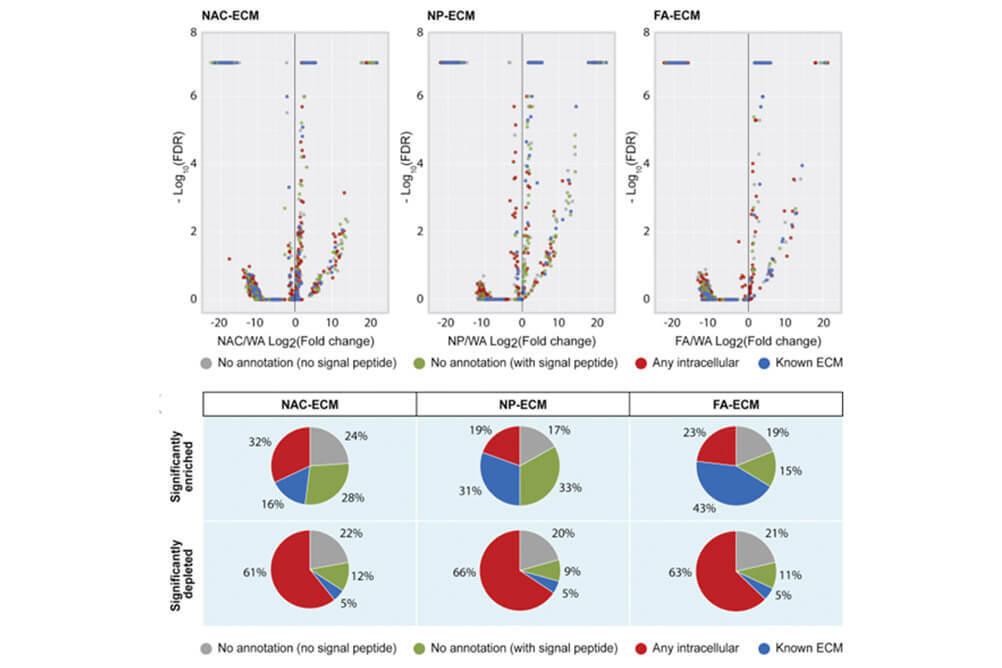
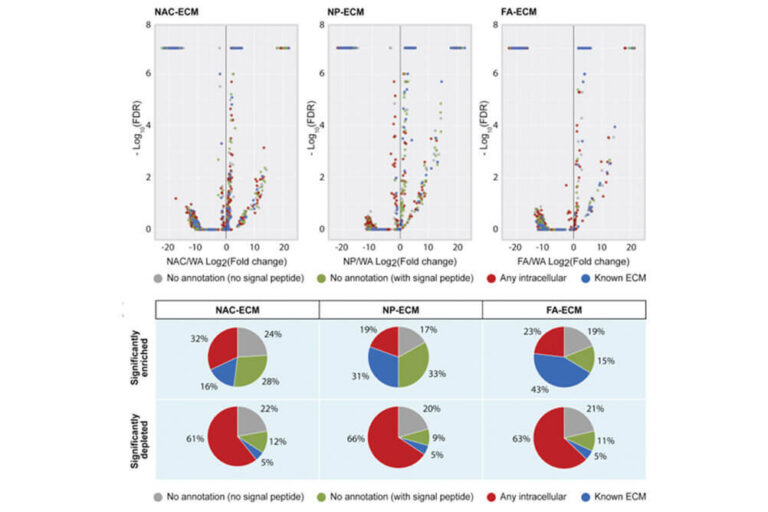
Decellularization enables functional analysis of ECM remodeling in planarian regeneration
Sonpho E, Mann FG, Jr., Levy M, Ross EJ, Guerrero-Hernandez C, Florens L, Saraf A, Doddihal V, Ounjai P, Sánchez Alvarado A. Mol Cell Proteomics. 2021:100137. doi: 100110.101016/j.mcpro.102021.100137.
